ABSTRACT
This review explores recent advances in the therapeutic potential of purine and its analogs, focusing on their development as promising agents against cancer, bacterial infections, viruses, fungi, parasites, and tuberculosis. With increasing resistance among pathogens, there is a critical need for novel antimicrobial agents to combat infectious diseases effectively. The review aims to deepen understanding by discussing both historical precedents and recent innovations in purine analogs and their derivatives as potent antimicrobial therapies. It highlights extensive research on various purine-containing compounds, demonstrating their efficacy in treating a wide spectrum of diseases and emphasizing the rapid progress in purine-based medicinal chemistry. Purine, characterized by a heterocyclic structure incorporating a fused pyrimidine and imidazole ring, offers diverse therapeutic properties. The review delves into structure-activity relationships and biological activities of promising purine molecules, positioning these compounds as leading candidates in drug development. The insights provided serve as a valuable resource for researchers engaged in synthesizing purine-based drug candidates, offering a roadmap for further exploration in this dynamic and evolving field.
Objectives:
- Evaluating the efficacy and safety of purine analogs in clinical and preclinical settings.
- Identifying structural features of purine analogs that correlate with their pharmacological activities.
- Discussing innovative approaches to overcome resistance to purine-based therapies. Keywords: Purine Analogs, Antimicrobial Agents, Therapeutic Potential, Drug Development, Structure-Activity Relationships
Introduction
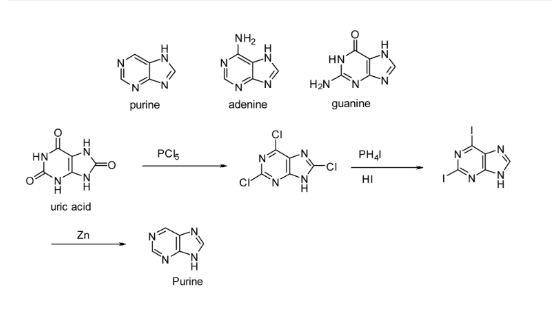
In the 1860s, Friedrich Miescher isolated "nuclein" from human pus cells, marking a significant milestone in nucleic acid chemistry. This material, composed of proteins and nucleic acids, laid the foundation for the field. The pivotal discovery of DNA's structure by Watson and Crick in 1953 further propelled scientific understanding [1]. Nucleic acids consist of a nucleobase (either purine or pyrimidine), a pentose sugar (D-ribose in RNA or deoxyribose in DNA), and phosphate groups [2]. Purine bases, namely guanine and adenine, contrast with pyrimidine bases like cytosine, thymine (in DNA), and uracil (in RNA). Purine, a fundamental nitrogenous base, was synthesized from uric acid by Emil Fischer in 1899 [3,4,5].
Essential purine bases such as adenine and guanine serve as enzyme cofactors (e.g., ATP, GTP, NAD, SAM, PAPS) and signaling molecules (e.g., adenosine, cAMP, cGMP), crucial for biological systems. Enzymes like nucleic acid polymerases, kinases, and nucleotide phosphorylases depend on purine derivatives for their functions.
Advancements in purine synthesis, driven by new synthetic techniques and technologies since Shaw et al.'s studies in 1996, have significantly contributed to pharmaceutical development in biology and medicine [6, 7]. Purine and its analogs have been extensively investigated for their potential as enzyme inhibitors (e.g., phosphodiesterases, p38a, Cdks, sulfotransferases, HSP90, MAP kinase, Src kinase, Clk inhibitors) [8–13], as well as for their antifungal [14], antibacterial [15], cytotoxic [16], antiviral [17], antihyperglycemic [18-20], and immunostimulatory properties [21-30].
Further literature has expanded on the broad biological actions of purine analogs, with recent focus on the years 2000–2020 by Wong et al. in medication development using nucleobase analogs [31-33]. Several nucleoside drugs approved by the FDA have shown promise against viruses such as SARS, Ebola, and Zika. Notably, remdesivir (RDV, GS-5734) has been highlighted for its efficacy in treating COVID-19 [34–38]. Ongoing clinical trials are evaluating drugs like valtorcitabine (anti-HBV) and elvucitabine (Fd4C; anti-HBV, anti-HIV). Novel purine modifications enabled by advancements in diazonium chemistry have also led to the discovery of nucleoside medicines [39–46].
This paper explores the recent advancement in cytotoxic Potential of Purine and its Analogs, emphasizing its role as a versatile agent against cancer, viruses, fungi, bacteria, parasites, and tuberculosis.
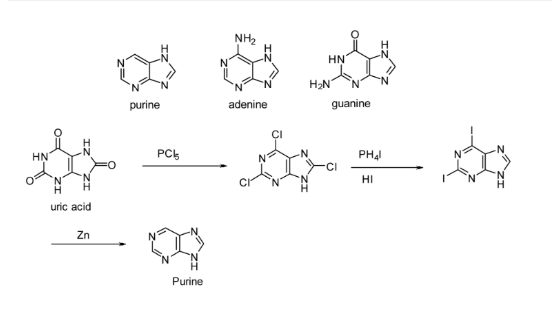
Figure 1. Synthesis of purine by E. Fischer [3].
Literature studies showed that tumor cell resistance to cytotoxic drugs is a major issue in cancer chemotherapy, stemming from intrinsic or acquired mechanisms. Multidrug resistance (MDR), linked to membrane P-glycoprotein overexpression, causes cross-resistance to drugs including vinca alkaloids, anthracyclines, podophyllotoxins, actinomycin D, taxol, and colchicine.
Systematic screening revealed that almitrine (1), a triazinylpiperazine used for respiratory insufficiency, moderately sensitized the highly resistant DC-3F/AD cell line to actinomycin D. This finding led to the synthesis of a series of analogs, resulting in the development of S9788 (2), now in Phase 1 clinical trials [47]. Preliminary clinical trials indicated the potential of S9788 in modulating resistance [48]. However, its cardiovascular side effects, such as prolonged QTc intervals, have limited its clinical use [49]. Consequently, achieving therapeutic plasma levels in patients has been difficult [50, 51]. Moreover, S9788 exhibits calcium channel affinity comparable to verapamil and significantly higher than almitrine [52,53]. Although long QT syndrome and Torsades de Pointes are not commonly associated with calcium channel blockers therapy [54], notable proarrhythmic effects have been seen with some calcium antagonists, such as bepridil, which prolong ventricular repolarization and QT intervals [55].
1. Anticancer Agents
Since the 1940s, significant strides have been made in medication development for cancer treatment [56-59] initially relying on nitrogen mustards and antifolate drugs. Recently, the FDA has approved an impressive array of 69 drugs and drug combinations tailored to combat various cancer types.
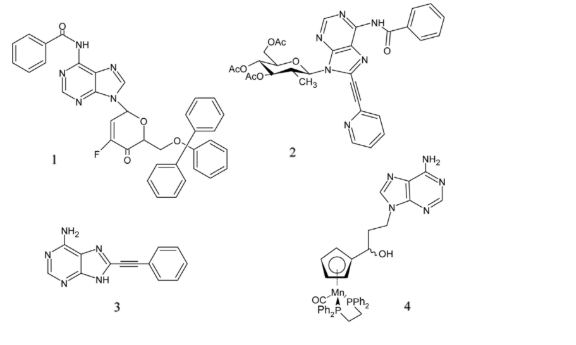
Current scientific research on adenine derivatives has yielded promising advancements in cancer therapy. Manta et al. [46] investigated the synthesis of a novel 3-fluoro-4-keto-D-glucopyranosyl derivative of N6-benzoyl adenine 1. This compound exhibited greater selectivity in inducing cytotoxic effects on several cancer cell lines, including MCF-7, Caco-2, and cutaneous melanoma, compared to 5-fluorouracil [60].
Dimopoulou et al. [61] identified two C8-alkynyl adenine derivatives that demonstrated novel anticancer properties against L1210, CEM, and HeLa cell lines, although they were found to be less effective than 5-fluorouracil.
Furthermore, Jablonski and colleagues [62] developed a highly effective antiproliferative agent in the form of a cymantrene-adenine derivative 4. Their research encompassed a diverse range of cancer cell lines including U-87-MG, HepG2, A549, SKOV-3, MDA-MB-231, MCF-7/DX, and MCF-7. Notably, this compound exhibited the highest degree of cytotoxicity among the tested cell lines, inducing oxidative stress that triggered autophagy and subsequent cell death in cancer cells.
These findings underscore the ongoing efforts to innovate cancer treatments through the development of adenine derivatives, highlighting their potential as potent agents in combating cancer cell proliferation and survival mechanisms.
Notably, adenine derivatives 5–10 were synthesized and studied for their anticancer effects on cancer cell lines LCLC-103H and HBC-5637. All of the derivatives had anticancer effects, according to the paper by Kamper et al. Compounds 6 and 7 in particular showed promising anticancer effects [63].
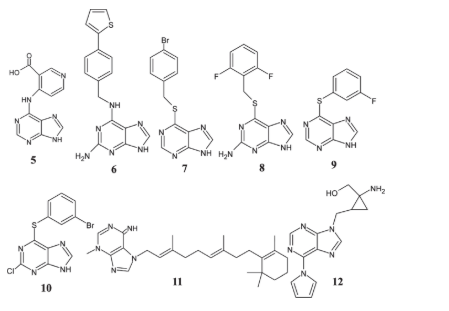
Agelasimine and agelasine are adenine derivatives that include β-cyclocitral. Analogues of these chemicals were developed and manufactured by Proszenyak and his coworkers. Several cancer cell lines were used to evaluate the chemicals. These included lymphoblastic lymphoma (RPMI 8226/s, U-937 GTB, and CEM/s) and renal cancer (ACHN) lines [64]. Amazingly, compound 11 showed higher anticancer activity than agelasimines and agelasines combined, and it beat traditional chemotherapy drugs like doxorubicin, paclitaxel, and cisplatin in terms of its antineoplastic characteristics. Also, Dzolic and coworkers demonstrated the synthesis of 1-amino-1-hydroxymethyl cyclopropane [65, 66]. When tested on a number of cell lines, including HeLa, Hep2, SW620, CEM, Molt4/C8, and FM3A, this purine analogue demonstrated significant anticancer and antiproliferative properties.
In a recent creative synthesis, Mohammed and colleagues [67] created derivatives 13–15 of tricyclic penciclovir and hydroxy butyl guanine. When studying these compounds, researchers employed a variety of human cancer cell lines, such as DND-41, hTERT RPE-1, Capan-1, NCI-H460, Hap1, Z-138, K-526, and HL-60. These derivatives nonetheless show promise despite having an alkyl group replaced on guanine's N9 and sharing similar chemical structures. Because of this, they might be pivotal in creating guanine derivative-based anticancer medications in the future.
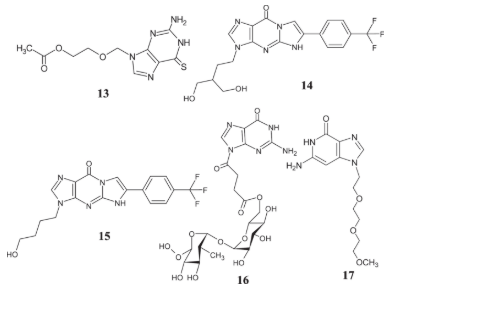
Analogs 16 and 17 of guanine developed by Musumeci et al. were more effective than the existing gold standard, cisplatin, in terms of efficacy [68]. These compounds were tested for their cytotoxic effects and ion transport capabilities using several human tumor cell lines, including SH-SY5Y, HaCaT, MCF-7, and DU145. Compound 16's anticancer efficacy outperformed the gold standard medicine in vitro, as shown by references [13–17].
1.1 Antitumor Agents
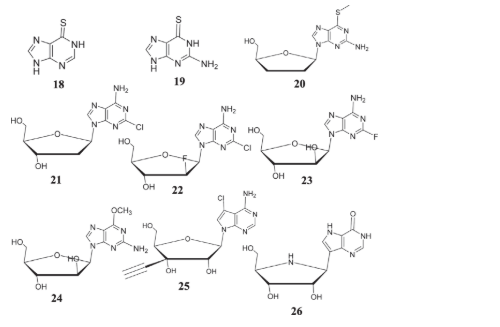
The relationship of purine and purine nucleoside structure to antitumor activity was reviewed by Robins in 1964 and by Goldin, Wood and Engle in 1968.[69,70,71, 72] Since that time several other useful reviews have appeared including a review by Montgomery in this journal in 1982 [73-77]. When it comes to treating leukemia and epithelial carcinoma, mercaptopurine (18) and mercaptoguanine (19) are recognised as effective. There is promising evidence that the zika virus may inhibit the activity of the furanose-associated chemical 20 [78]. When it comes to leukemia and lymphoma, halogenated nucleosides like cladribine (21), clarabine (22), and fludarabine (23), among others, are miracle workers. With its 9-β-D-arabinose furan and guanine derivative, the water-soluble prodrug nairadbine effectively fights T-cell leukemia (T-ALL) and T-cell malignant tumors (T-LBL). In 2006, the FDA approved this chemical due to its improved therapeutic benefits [79]. Using the 30-ethynyl iso-nucleoside to suppress metastatic breast tumors has been shown to be effective [59]. One guanine analog that has shown promise in treating leukemia, lymphocytic leukemia, Hodgkin's lymphoma, and skin cancers linked to T cells is forodesine (26), which has been authorized by the FDA [80].
1.2 Inhibitors of Hsp90
Hsp90 plays a crucial role in refolding damaged or misfolded proteins and aiding in the structural development of many oncogenic signaling proteins. In the absence of Hsp90, these proteins would be broken down by the proteasome. Thus, a novel method for treating cancer involves blocking Hsp90, which in turn triggers the proteasome to degrade these proteins [31, 81-85].
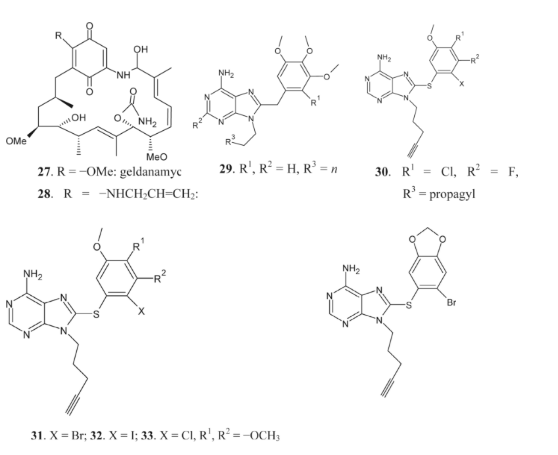
The ansamycins geldanamycin (AAG) 27 and 17-AAG 28 were identified as the first inhibitors of Hsp90. This is due to the fact that these ansamycins bind more strongly to the N-terminal pocket of Hsp90 in tumor cells than in normal cells. Also, as stated before, Hsp90 is efficiently blocked by purine derivatives 29 and 30 [86, 87]. Compound 34 inhibits Hsp90 with the best selectivity and efficacy among the purine-based compounds, according to results from research into the structure-activity relationship and synthesis of 8-arylsulfonyl adenine derivatives [31]. Hsp90, which triggers cell cycle arrest and death in cancer cells, is an important target for synthetic inhibitors. Potentially helpful in cancer therapy, Hsp90 inhibitors block the action of many carcinogenic proteins simultaneously** [88, 89]**.
1.3 Inhibitors of Microtubule Assembly
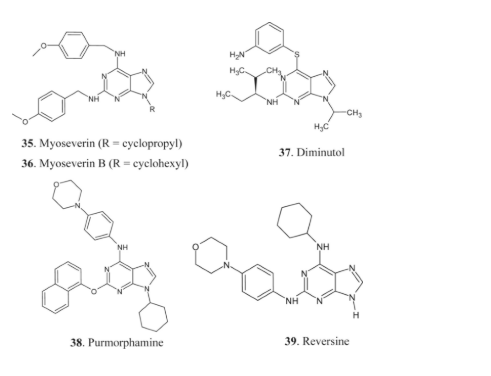
Chang and coworkers found myoseverin 35, a powerful inhibitor of microtubules. By preventing spindle formation, it halts the cell cycle during the G2/M transition stages [67]. The low toxicity of myoseverin and its derivatives makes them attractive cytostatic anticancer medication candidates. Researchers are looking at a number of chemicals, including myoseverin B, a N9-cyclohexyl derivative, for potential anticancer therapeutic properties. A few other examples include reversine, diminutol, and purmorphamine [31, 32, 90-96].
1.4 Inhibitors of Src Tyrosine Kinase
When certain cell receptors are activated, Src tyrosine kinase works as an early signal transducer [85, 86]. There are a number of chemical families that can compete with Src kinase for the binding site of ATP and hence decrease its activity [65, 66].
Osteosarcoma patients are now being treated with a family of Src inhibitors known as purine analogs 40-43 [97, 98]. An outstanding purine molecule with promising therapeutic potential against chronic myelogenous leukemia is phoshonite oxide 43. The IC50 value is less than 1 nM, making it highly selective and strong in inhibiting both Abl kinases and Src. In addition to acute myelogenous leukemia and systemic mastocytosis, this treatment may be useful in future clinical studies [31, 99,100-103].
Inhibitors of Cyclin-Dependent Kinase
Essential for cell proliferation, cyclin-dependent kinases (Cdks) regulate many steps of the cell cycle. Cdk inhibitors have the ability to induce cell death in tumor cells by interfering with their G1 and G2 phases [104, 105].
There has been a lot of interest in the synthesis of 2,6,9-trisubstituted purines as Cdk5, Cdk2, and Cdk51 specific inhibitors. Pharmaceuticals such as roscovitine (45) and olomoucine (44) have been significant in advancing this area. Among the most powerful inhibitors, purvalanol A (46), stands out; nonetheless, its pharmacological profile is more limited than that of the orally administered roscovitine (CYC202 or seliciclib, 45). Frequently used in conjunction with cisplatin or gemcitabine, roscovitine is now being studied in phase II clinical trials for hematological malignancies, multiple myeloma, lung cancer, and multiple myeloma [31, 92, 93].
An exciting new direction in the search for selective and effective cancer treatments is the targeting of Cdks. In addition to parasite illnesses including Plasmodium, Trypanosoma, and Leishmania, these inhibitors show promise against a variety of viruses including herpes, HPV, HIV, and CMV [75, 106, 107].
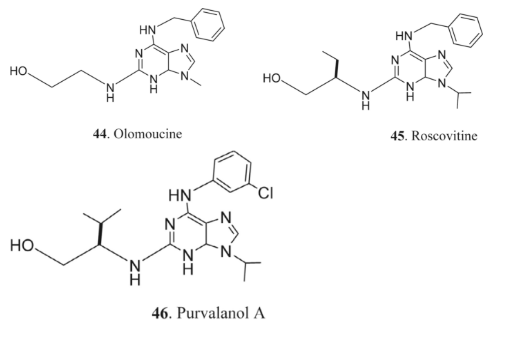
2 Antiparasitic Agents
Literature provide wide variety of purine analogs as Antiparasitic Agents [108-115]. Jablonski and colleagues have highlighted the potential of cymantrenyl-adenine derivatives as effective antitrypanosomal drugs, in addition to their known antibacterial properties [50]. Among these derivatives, compound 4 has demonstrated superior efficacy against T. brucei compared to uracil-based compound 13. The enhanced effectiveness of compound 4 is attributed to its lipophilic phosphine ligands [46].
Similarly, agelasimine analogs, a specific class of adenine derivatives, have shown promising antiparasitic effects. Compounds 47 and 48 have exhibited significant activity against A. polyphaga and A. castellanii, respectively [52]. These findings underscore the potential of adenine derivatives in developing new therapies against parasitic infections, suggesting avenues for further exploration in antiparasitic drug development.
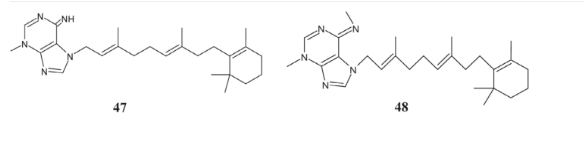
3 Antifungal Agents
The proliferation of diverse fungal strains has significantly contributed to the alarming rise in severe fungal infections, highlighting the critical need for innovative antifungal medications. While fungal kinases are often shared with higher eukaryotic organisms, fungi utilize these enzymes to regulate essential processes such as the cell cycle. A promising approach in antifungal drug development involves targeting specific, nonhomologous kinases, such as histidine kinase, which are distinct to fungi [116].
Research by Zaidan and Mohamed has demonstrated that adenine derivatives containing sulfur and azo groups possess potent antifungal properties [117]. Specifically, compound 49 has shown increased efficacy against A. flavus. The antifungal activity of this compound is attributed to its structural composition, which includes azo groups and sulfur atoms within its sulfonic acid structure [118, 119]. These findings suggest a pathway for developing novel antifungal agents that could effectively combat a wide range of fungal infections by targeting specific biochemical pathways unique to fungi.
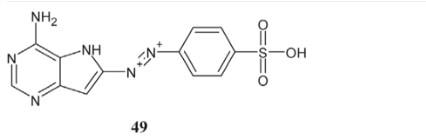
The synthesis of 6-amino aromatic-2-cyclohexyl diamino purine derivatives (50a, b, c, d, e; R = H, ET, Me, cyclopentyl, iPr) as efficient inhibitors of fungal CaCiv1 and CAK was recorded by Bordon-Pallier and their colleagues [18]. Research is now focused on improving the effectiveness of these inhibitors and understanding how they bind to the active sites of these unique kinases.
4 Antibacterial Agents
A restricted fraction of nucleobase analog drugs (NADs) have shown therapeutic advantages against bacterial infections, limiting their usefulness at this time. People aren't very interested in creating antimicrobial medications that can fight the fast-growing population of bacteria that are resistant to antibiotics, thus these chemicals are mostly used as cancer and virus treatments [120].
The antibacterial activities of cymantrenyl adenine derivatives were first discovered by Jablonski and colleagues [50]. Compound 4 has modest antibacterial action against MSSA, VISA, S. aureus, S. epidermidis, and other Gram-positive bacteria.
Zaidan and Mohamed published a different investigation on azo-and sulfur-containing adenine derivatives, drawing attention to compound 50's antibacterial activity against Staphylococcus aureus and Escherichia coli [121]. Furthermore, compound 51 of the agelasimine analogs studied by Proszenyák et al., which were initially created to combat cancer, showed substantial efficacy against Mycobacterium TB, Staphylococcus aureus, and Escherichia coli** [52]**. The urgent problem of antibiotic resistance in microbial infections is being addressed by continuing attempts to investigate new adenine derivatives with antibacterial characteristics. To combat new and emerging bacterial infections, this field urgently needs further research and development.
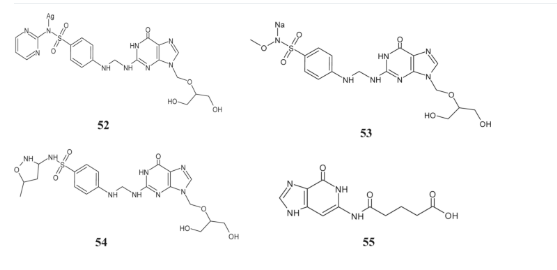
Joshi et al. developed new guanine derivatives, specifically Mannich bases of ganciclovir, which have exhibited potent antibacterial capabilities [122]. Their research focused on evaluating the antimicrobial activity of these compounds (52–55) against a spectrum of bacteria, including Staphylococcus aureus, Escherichia coli, and Bacillus subtilis.
The effectiveness of these derivatives was assessed using the cup plate technique, comparing them to conventional sulfonamide drugs. Results demonstrated that compounds 52–55 exhibited significantly higher activity levels compared to standard sulfonamides, effectively inhibiting the growth of bacterial strains to a considerable extent. These findings highlight the potential of these new guanine derivatives as promising candidates for further development in combating bacterial infections.
Microbial infections have become increasingly prevalent in recent decades compared to the early 1900s, posing ongoing challenges in diagnosis and treatment. In clinical practice, antibacterial medications, whether synthetic or semi-synthetic, have played a crucial role [123]. Despite significant advancements in antimicrobial treatment, many currently available drugs still face certain limitations. Therefore, there remains a critical need to discover and develop novel antimicrobial medicines that offer improved pharmacological characteristics. This pursuit is essential to effectively combatting evolving microbial threats and enhancing treatment outcomes in clinical settings.
5. Antitubercular Agents
As new drug-resistant tuberculosis strains arise, the illness continues to pose a serious threat to public health. There are effective antitubercular drugs available, but there is a constant need for fresh research and development due to the emergence of drug-resistant forms. Preliminary investigations have shown promise for several 6-thio-N9-substituted purine derivatives, which are among the promising options [124, 125].
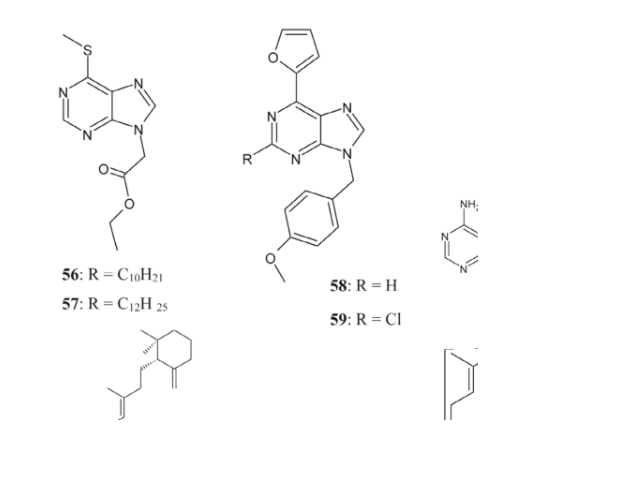
Particularly noteworthy were compounds 56 and 57, which demonstrated antitubercular activity. The minimum inhibitory concentrations (MICs) against the Mtb H37Rv strain were 1.56 μg/mL and 0.78 μg/mL, respectively, for compound 56, also known as (6-decyl sulfanyl-purin-9-yl)-acetic acid ethyl ester, and its dodecyl analog, 57 [33, 126].
Compounds 58 and 59, on the other hand, showed substantial efficacy against Mycobacterium tuberculosis with minimum inhibitory concentration (MIC) values of 1.56 μg/mL and 0.39 μg/mL, respectively, without being as hazardous to adult stem cells** [33, 127]. We need to do additional research to determine the exact mechanisms of action for these drugs.
Also, an extract from sea sponges called Agelasine F (60) has been shown to be effective against Mycobacterium tuberculosis. Comparable antitubercular actions have been shown by analogous compounds, namely 61 and 62, which are 7,9-dialkyl purinium salts **[31, 33, 128]. These results highlight the possibility that related chemicals and purine derivatives with 6-thio-N9 substitution might be effective in combating drug-resistant TB.
5 Antiviral Agents
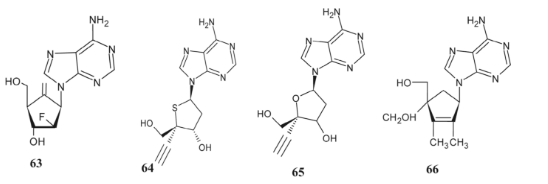
Notable antiviral activity has been shown by many adenine derivatives that feature a 5-membered ring substituent. Using an intracellular HBV DNA replication assay, Wang and colleagues [90] evaluated the efficacy of 2′-fluoro-6′-methyl carbocyclic adenosine (63), a novel medication. Another molecule, 65, was shown to be less efficient against HIV-1, while 4′-Thioadenosine (64) was discovered by Haraguchi and colleagues [129] as having potential antiviral activity.
However, compound 64 greatly reduced the activity of vaccinia virus, herpes simplex virus type 1, and herpes simplex virus type 2. To combat viruses, Ko and colleagues **[130] **created novel adenine-based 2′,3′,4′-triply branched carbocyclic nucleosides. Comparing these compounds to the current standard of care, ribavirin, compound 66 showed limited efficacy against human cytomegalovirus.
All things considered, adenine derivatives 63–66, which have a 5-membered ring substituent, showed substantial antiviral activity. The sulfur-containing chemical 64 and the oxygen-containing compound 65 were the most powerful of the bunch.
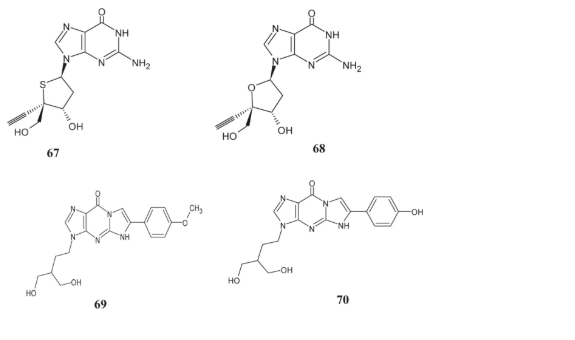
The antiviral characteristics of a 4′-ethynyl-2′-deoxy-4′-thio ribonucleoside derivative were discovered by Haraguchi et al. [117]. The activity of 4′-thioguanine (67) in HIV-1 suppression tests was lower than that of the related 4′-ethynyl derivatives of 2-deoxyguanosine (68). In HEL cell cultures, Compound 69 showed potent inhibitory effects against HSV-1 (KOS, TK-ACV), HSV-2 (G), and vaccinia virus; nevertheless, it did not achieve the same level of efficacy as the gold standard antiviral drug, ganciclovir.
The team led by Mohammed also revealed the creation of tricyclic penciclovir compounds and hydroxy butyl guanine, both of which exhibited potent antiviral effects [55]. When it came to HSV-1 and HSV-2, compound 75 stood out among the others. Compound 70 showed strong inhibitory effects against the thymidine kinase positive varicella zoster virus and successfully targeted the HSV-1 strain that is resistant to TK-ACV. In addition, compound 69 demonstrated superior efficacy compared to acyclovir, cidofovir, and brivudine, which are regularly used antiviral drugs.
The efficiency of these compounds is dependent on a dihydroxyl acyclic sugar moiety, according to structure-activity relationship (SAR) research [33].
6.1. Interferon Inducers as Antiviral Agents
Adenine derivatives have demonstrated significant potential as inducers of interferon (IFN), presenting a compelling class of drugs for treating viral infections. IFNs are antiviral proteins that constitute one of the body's primary defenses against viruses. Their capability to modulate immune responses has also positioned them as a promising treatment option for various cancers and autoimmune diseases [130].
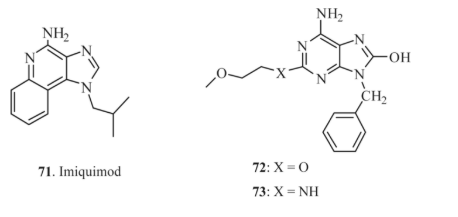
Modern treatment for chronic hepatitis C often involves the use of interferon-α, either alone or in combination with ribavirin. However, the presence of antibodies that neutralize the administered exogenous interferon typically reduces the efficacy of this therapy. To overcome this limitation, orally administered small-molecule drugs, such as imiquimod 71, have been developed to stimulate the production of endogenous interferon (IFN). It represents a novel approach in combating viral infections [131-136].
Research on mice, at a dose of 1 mg/kg, demonstrated that purine derivatives 72 and 73 were more effective and bioavailable than imiquimod, with IFN activation levels of 2171 IU/mL and 974 IU/mL, respectively [31,137 , 138].
Hirota and colleagues [120] focused on developing purine derivatives capable of inducing IFN production. They synthesized 2-substituted-benzyl-8-dihydroxyadenine derivatives 74–78 (R = –SCH3, –SC2H5, –SC3H7, –NHC4H9, –OC4H9), which proved to be more effective in eliciting IFN production than imiquimod.
6.2. Adenine Nucleoside Analogs
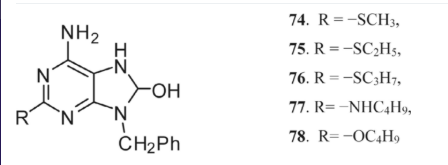
The nucleoside reverse transcriptase inhibitor tenofovir 79 is an HIV medicine [139-146]. Its low GI absorption rate, however, prompted the creation of a number of prodrugs. Treatment of hepatitis B virus infection makes use of prodrugs such as tenofovir disoproxil fumarate 80 and adefovir dipivoxil 81, which are resistant to HIV. Side effects include renal failure and lactic acidosis are linked to these drugs when used for an extended period of time.
Researchers have created pradefovir 82, a prodrug of adefovir phosphonate with a 4-aryl cyclic ring, to deal with the problem of kidney toxicity. The antiviral efficacy, resistance profile, and decreased nephrotoxicity of this medication are better than those of adefovir dipivoxil 81 [35, 147]. Furthermore, alamifovir 83 is now undergoing clinical studies. Alamifovir has greater action against HBV and is more effective than lamivudine [34, 148].
6.3. Guanine Nucleoside Analogs
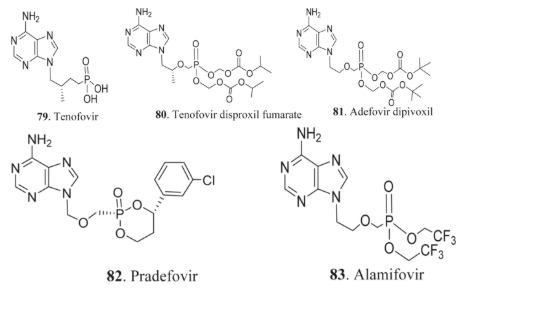
Herpes infections can present in various forms, affecting different parts of the body. Medications such as acyclovir 84, a guanine nucleoside analog, are commonly used to treat conditions like chickenpox, genital herpes, and shingles, which can cause damage to the skin, brain, and oral mucosa [149-153]. Through ongoing modifications to the sugar chain in the nucleoside structure, additional compounds such as ganciclovir 85, penciclovir 86, famciclovir 87, valacyclovir 88, and valganciclovir 89 have been developed. These compounds exhibit lower toxicity and improved bioavailability, making them effective treatments for herpes and cytomegalovirus (CMV) infections. Oral administration significantly enhances the efficacy of compounds 88-90.
Another notable prodrug with a broad spectrum of antiviral activity is cycloganciclovir 90, which functions in a manner similar to cyclic GMP (cGMP) [35, 111]
6.4. RNA-Dependent RNA Polymerase Inhibitors
Because of their lipophilicity, one of the main obstacles to improving the effectiveness of anti-HIV drugs is getting them to cross the blood-brain barrier. Modifications at the purine 6-position are often used to address this problem [154-158]. While 6-methylmercaptopurine riboside 20 and 20-F-6-ethylamino adenosine 91 and 20-F-6-dimethylamino adenosine 92, the prodrugs' unmodified parent molecules, are less lipophilic than diadenosine or its 20-F analog, prodrugs like these effectively suppress the Zika virus [159].
When it comes to treating HIV, the nucleoside 20-F-ara-ddA is aimed squarely at the central nervous system. The therapeutic utility of 20-F-ara-ddA is enhanced by its prodrug, 20-F-6-Azido adenosine 93 [160].
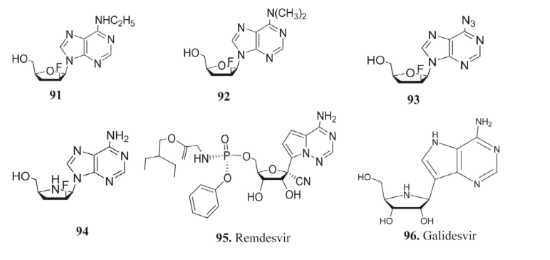
Although it does not work against HIV, adenine analog 94 shows that it can neutralize a wide range of other RNA viruses [57]. Remdesivir 95, a broad-spectrum antiviral prodrug based on adenosine, was the focus of much study during the COVID-19 pandemic because of its efficacy against SARS-CoV and MERS [99, 100]. One of the most important enzymes in viral RNA replication, RNA-dependent RNA polymerase (RdRp) [40], is inhibited by Remdesivir.
Remdesivir has distinctive resistance to COVID-19 exonuclease, which is one of its distinguishing properties. Although this enzyme normally breaks down nucleoside analogs that are part of viral RNA, Remdesivir's resistance makes it a more effective barrier against COVID-19 than other nucleoside analogs** [34, 161-165].
Galidesivir (BCX4430) 96, an adenosine nucleoside analog, has also shown enhanced antiviral effectiveness against a number of virus families, including those that cause yellow fever, Marburg, Zika, Ebola, and Rift Valley sickness. Galidesivir has shown more effectiveness in animal models than other cell line cultures, and phase I clinical studies are already under progress. Evasion tests against SARS coronavirus 2 and Marburg virus are also a part of this continuing research **[166-170].
Future Perspective
Over the years, and more recently, purine-based drugs have been extensively employed to address a broad spectrum of medical conditions, including diabetes, cancer, rheumatoid arthritis, osteoporosis, asthma, and depression. The FDA has approved several cytotoxic drugs derived from purine analogs. These include adenine analogs such as fludarabine, clofarabine, cladribine, mercaptopurine, vidarabine, didanosine, adefovir, tenofovir, agelasines, and agelasimines, as well as guanine analogs like thioguanine, mercaptopurine, nelarabine, penciclovir, famciclovir, entecavir, valganciclovir, valacyclovir, abacavir, and azathioprine (a prodrug of mercaptopurine). These compounds are integral to various therapeutic regimens.
Despite the extensive use of these analogs, the emergence of microbial resistance and the continuous spread of infectious diseases necessitate innovative strategies involving cytotoxic agents derived from synthetic and semi-synthetic purine scaffolds. For the development of novel therapeutic agents, it is crucial to explore the pharmacological relationships between enzyme inhibition and cytotoxic efficacy.
Compared to adenine analogs, guanine analogs are less represented in the literature as therapeutic agents. This discrepancy has captured the attention of medicinal chemists, who are now investigating guanine analogs as potential new therapeutic targets. Purine, with its multiple reactive sites, exhibits extensive pharmacological activity, particularly in N9-substituted derivatives. However, there is a growing interest in exploring the cytotoxic effects of tautomeric N7-substituted purine analogs and other reactive centers in substituted purine entities.
To effectively investigate these cytotoxic effects, medicinal chemists are encouraged to employ advanced techniques such as high-throughput screening, microwave-assisted synthesis, molecular modeling, and structure-activity relationship (SAR), quantitative structure-activity relationship (QSAR), and quantitative structure-property relationship (QSPR) studies. These modern methodologies can significantly enhance the understanding of the pharmacological potential of purine derivatives.
Furthermore, improving the bioavailability, pharmacological properties, and membrane permeability of these prodrugs holds the promise of developing new clinical therapeutic agents. Such advancements could push the boundaries of drug design and yield effective treatments for a wide range of disorders. The ongoing exploration of purine analogs underscores the dynamic nature of medicinal chemistry and its potential to address pressing healthcare challenges through innovative drug design.
Future Perspective
The field of purine analog research is at a critical juncture, marked by significant advances and ongoing challenges. As resistance to conventional therapeutics continues to evolve, the necessity for innovative approaches in drug development becomes increasingly imperative. Future research should focus on several key areas:
- Enhanced Selectivity and Potency: Developing purine analogs with improved selectivity for specific cellular targets will minimize side effects and enhance therapeutic efficacy. The use of high-throughput screening and computational modeling to predict interaction dynamics can aid in the design of molecules with precise action.
- **Combination Therapies: **Exploring the synergistic effects of purine analogs combined with other therapeutic agents could offer new treatment protocols, particularly in cancer and infectious diseases, to overcome resistance mechanisms and achieve better clinical outcomes.
- Novel Delivery Systems: Enhancing the bioavailability and delivery of purine analogs through innovative formulations such as nanoparticle systems or targeted delivery mechanisms can improve their efficacy and reduce toxicity.
- Expanded Spectrum of Diseases: Although purine analogs are extensively used in cancer and antiviral therapies, their potential in treating autoimmune diseases, neurological disorders, and metabolic syndromes remains underexplored. Research should be directed towards these areas to broaden the therapeutic spectrum of purine derivatives.
- Tackling Drug Resistance: With the persistent emergence of drug-resistant strains, especially in microbial pathogens, there is an urgent need to understand resistance mechanisms and develop purine analogs that can circumvent or overcome these barriers.
References
Strauss, B. S. (2018). Why is DNA double stranded? The discovery of DNA excision repair mechanisms. Genetics, 209(2), 357-366.
Saenger, W. (2013). Principles of nucleic acid structure. Springer Science & Business Media.
Fischer, E. (1899). Purine synthesis. Berichte der Deutschen Chemischen Gesellschaft, 32, 2550.
Porter, A. E. A. (1984). The structure, reactions, synthesis, and uses of heterocyclic compounds. Comprehensive Heterocyclic Chemistry, 3, 157-197..
Chang, Y. T., Gray, N. S., Rosania, G. R., Sutherlin, D. P., Kwon, S., Norman, T. C., ... & Schultz, P. G. (1999). Synthesis and application of functionally diverse 2, 6, 9-trisubstituted purine libraries as CDK inhibitors. Chemistry & biology, 6(6), 361-375.
Wang, C., Song, Z., Yu, H., Liu, K., & Ma, X. (2015). Adenine: an important drug scaffold for the design of antiviral agents. Acta pharmaceutica sinica B, 5(5), 431-441.
Haystead, J., & Timothy, A. (2006). The purinome, a complex mix of drug and toxicity targets. Current topics in medicinal chemistry, 6(11), 1117-1127.
MORIGUCHI, I., & KANADA, Y. (1977). Use of van der Waals volume in structure-activity studies. Chemical and Pharmaceutical Bulletin, 25(5), 926-935.
Bråthe, A., Andresen, G., Gundersen, L. L., Malterud, K. E., & Rise, F. (2002). Antioxidant activity of synthetic cytokinin analogues: 6-alkynyl-and 6-alkenylpurines as novel 15-lipoxygenase inhibitors. Bioorganic & medicinal chemistry, 10(5), 1581-1586.
Bråthe, A., Gundersen, L. L., Malterud, K. E., & Rise, F. (2005). 6‐Substituted Purines as Inhibitors of 15‐Lipoxygenase; a Structure‐Activity Study. Archiv der Pharmazie: An International Journal Pharmaceutical and Medicinal Chemistry, 338(4), 159-166.
Laufer, S. A., Domeyer, D. M., Scior, T. R., Albrecht, W., & Hauser, D. R. (2005). Synthesis and biological testing of purine derivatives as potential ATP-competitive kinase inhibitors. Journal of medicinal chemistry, 48(3), 710-722.
Qin, Z., Qin, L., Feng, X., Li, Z., & Bian, J. (2021). Development of Cdc2-like kinase 2 inhibitors: achievements and future directions. Journal of medicinal chemistry, 64(18), 13191-13211.
Das, S. K., Behera, B., & Purohit, C. S. (2023). Scaffolds of Purine Privilege for Biological Cytotoxic Targets: A Review. Pharmaceutical Chemistry Journal, 57(6), 777-792.
Peterson, E. M., Brownell, J., & Vince, R. (1992). Synthesis and biological evaluation of 5'-sulfamoylated purinyl carbocyclic nucleosides. Journal of medicinal chemistry, 35(22), 3991-4000.
Gangjee, A., Vasudevan, A., & Queener, S. F. (1997). Conformationally restricted analogues of trimethoprim: 2, 6-diamino-8-substituted purines as potential dihydrofolate reductase inhibitors from Pneumocystis carinii and Toxoplasma gondii. Journal of medicinal chemistry, 40(19), 3032-3039.
Gumina, G. and Chu, C.K., 2002. Synthesis of L-oxetanocin. Organic Letters, 4(7), pp.1147-1149.
Das, S.K., Behera, B. and Purohit, C.S., 2023. Scaffolds of Purine Privilege for Biological Cytotoxic Targets: A Review. Pharmaceutical Chemistry Journal, 57(6), pp.777-792.
Bordon-Pallier, F., Jullian, N., Ferrari, P., Girard, A.M., Bocquel, M.T., Biton, J., Bouquin, N. and Haesslein, J.L., 2004. Inhibitors of Civ1 kinase belonging to 6-aminoaromatic-2-cyclohexyldiamino purine series as potent anti-fungal compounds. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1697(1-2), pp.211-223.
Patel, P.R., Ramalingan, C. and Park, Y.T., 2007. Synthesis and antimicrobial evaluation of guanylsulfonamides. Bioorganic & medicinal chemistry letters, 17(23), pp.6610-6614.
Thomas, H.J. and Montgomery, J.A., 1968. Derivatives and analogs of 6-mercaptopurine ribonucleotide. Journal of Medicinal Chemistry, 11(1), pp.44-48.
Andresen, G., Gundersen, L.L., Nissen-Meyer, J., Rise, F. and Spilsberg, B., 2002. Cytotoxic and antibacterial activity of 2-oxopurine derivatives. Bioorganic & medicinal chemistry letters, 12(4), pp.567-569.
Wang, J.F., Zhang, L.R., Yang, Z.J. and Zhang, L.H., 2004. Synthesis and biological evaluation of 6-substituted purine and 9-β-D-ribofuranosyl purine analogues. Bioorganic & medicinal chemistry, 12(6), pp.1425-1429.
Hocek, M., Nauš, P., Pohl, R., Votruba, I., Furman, P. A., Tharnish, P. M., & Otto, M. J. (2005). Cytostatic 6-arylpurine nucleosides. 6. SAR in anti-HCV and cytostatic activity of extended series of 6-hetarylpurine ribonucleosides. Journal of medicinal chemistry, 48(18), 5869-5873.
Sidwell, R. W., Huffman, J. H., Khare, Lois, G. P., Allen, B., Witkowski, Roland, J. T., & Robins, K. (1972). Broad-spectrum antiviral activity of virazole: 1-β-D-ribofuranosyl-1, 2, 4-triazole-3-carboxamide. Science, 177(4050), 705-706.
De Clercq, E. (1987). S-Adenosylhomocysteine hydrolase inhibitors as broad-spectrum antiviral agents. Biochemical pharmacology, 36(16), 2567-2575.
Clerq, E. D. (1998). Carbocyclic adenosine analogues as S-adenosylhomocysteine hydrolase inhibitors and antiviral agents: recent advances. Nucleosides & Nucleotides, 17(1-3), 625-634.
De Clercq, E. (2005). Antiviral drug discovery and development: where chemistry meets with biomedicine. Antiviral research, 67(2), 56-75.
Jin, G., Wu, C. C., Tawatao, R. I., Chan, M., Carson, D. A., & Cottam, H. B. (2006). Synthesis and immunostimulatory activity of 8-substituted amino 9-benzyladenines as potent Toll-like receptor 7 agonists. Bioorganic & medicinal chemistry letters, 16(17), 4559-4563.
Legraverend, M., & Grierson, D. S. (2006). The purines: Potent and versatile small molecule inhibitors and modulators of key biological targets. Bioorganic & medicinal chemistry, 14(12), 3987-4006.
Haystead, J., & Timothy, A. (2006). The purinome, a complex mix of drug and toxicity targets. Current topics in medicinal chemistry, 6(11), 1117-1127.
Wong, X. K., & Yeong, K. Y. (2021). From Nucleic Acids to Drug Discovery: Nucleobases as Emerging Templates for Drug Candidates. Current Medicinal Chemistry, 28(34), 7076-7121.
Lin, X., Liang, C., Zou, L., Yin, Y., Wang, J., Chen, D., & Lan, W. (2021). Advance of structural modification of nucleosides scaffold. European Journal of Medicinal Chemistry, 214, 113233.
Lo, M. K., Feldmann, F., Gary, J. M., Jordan, R., Bannister, R., Cronin, J., ... & de Wit, E. (2019). Remdesivir (GS-5734) protects African green monkeys from Nipah virus challenge. Science translational medicine, 11(494), eaau9242.E. P. Tchesnokov, J. Y. Feng, D. P. Porter, et al., Viruses, 11, 326 (2019).
Tchesnokov, E. P., Feng, J. Y., Porter, D. P., & Götte, M. (2019). Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses, 11(4), 326.
Sheahan, T. P., Sims, A. C., Leist, S. R., Schäfer, A., Won, J., Brown, A. J., ... & Baric, R. S. (2020). Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nature communications, 11(1), 222.
Wang, M., Cao, R., Zhang, L., Yang, X., Liu, J., Xu, M., ... & Xiao, G. (2020). Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell research, 30(3), 269-271.
Ikejiri, M., Saijo, M., Morikawa, S., Fukushi, S., Mizutani, T., Kurane, I., & Maruyama, T. (2007). Synthesis and biological evaluation of nucleoside analogues having 6-chloropurine as anti-SARS-CoV agents. Bioorganic & medicinal chemistry letters, 17(9), 2470-2473.
Taylor, R., Kotian, P., Warren, T., Panchal, R., Bavari, S., Julander, J., ... & Sheridan, W. P. (2016). BCX4430–a broad-spectrum antiviral adenosine nucleoside analog under development for the treatment of Ebola virus disease. Journal of infection and public health, 9(3), 220-226.
Chen, Y. L., Yokokawa, F., & Shi, P. Y. (2015). The search for nucleoside/nucleotide analog inhibitors of dengue virus. Antiviral research, 122, 12-19.
Bernatchez, J. A., Coste, M., Beck, S., Wells, G. A., Luna, L. A., Clark, A. E., ... & Siqueira-Neto, J. L. (2019). Activity of selected nucleoside analogue protides against zika virus in human neural stem cells. Viruses, 11(4), 365.
DiRocco, D. A., Ji, Y., Sherer, E. C., Klapars, A., Reibarkh, M., Dropinski, J., ... & Davies, I. W. (2017). A multifunctional catalyst that stereoselectively assembles prodrugs. Science, 356(6336), 426-430.
Seley-Radtke, K. L., & Yates, M. K. (2018). The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral research, 154, 66-86.
Yates, M. K., & Seley-Radtke, K. L. (2019). The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antiviral research, 162, 5-21.
Sengupta, S., & Das, P. (2022). Application of diazonium chemistry in purine modifications: A focused review. Journal of Heterocyclic Chemistry, 59(1), 5-21.
Bhutani, P., Joshi, G., Raja, N., Bachhav, N., Rajanna, P. K., Bhutani, H., ... & Kumar, R. (2021). US FDA approved drugs from 2015–June 2020: a perspective. Journal of Medicinal Chemistry, 64(5), 2339-2381.
Manta, S., Agelis, G., Botić, T., Cencič, A., & Komiotis, D. (2008). Unsaturated fluoro-ketopyranosyl nucleosides: Synthesis and biological evaluation of 3-fluoro-4-keto-β-D-glucopyranosyl derivatives of N4-benzoyl cytosine and N6-benzoyl adenine. European journal of medicinal chemistry, 43(2), 420-428.
Dhainaut, A., Régnier, G., Atassi, G., Pierré, A., Léonce, S., Kraus-Berthier, L., & Prost, J. F. (1992). New triazine derivatives as potent modulators of multidrug resistance. Journal of Medicinal Chemistry, 35(13), 2481-2496.
Terret, C., Goncalves, E., Ruffie, P., Sarkany, M., Giroux, B., Lucas, C., Funck-Brentano, C., & Armand, J. P. (1996). Specific methodology for a phase I trial with unusual side-effect: S9788. 9th NCI-EORTC Symposium on New Drugs in Cancer Therapy, Abstract 438.
El-Sherif, N. (1991). Early afterdepolarizations and arrhythmogenesis: Experimental and clinical aspects. Archives des Maladies du Coeur et des Vaisseaux, 84(2), 227-234.
Franz, M. R., Koller, B., & Woosley, R. L. (1994). Pharmacologic therapy in cardiac arrhythmias. In N. Bramah Singh, V. J. Dzau, P. M. Vanhoutte, & R. L. Woosley (Eds.), Cardiovascular Pharmacology and Therapeutics (pp. 595-616). Edinburgh, U.K.: Churchill Livingstone Inc.
Jaillon, P., & Simon, T. (1992). Les effets pro-arythmiques ventriculaires des médicaments antiarythmiques. (Ventricular proarrhythmic effects of antiarrhythmic drugs). Thérapie, 47, 187-192.
Somberg, J. C. (1985). Calcium channel blockers that prolong the QT interval. American Heart Journal, 110(2), 416-421.
Mitsunobu, O. (1981). The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis, 1981(1), 1-28.
Kazimierczuk, Z., Cottam, H. B., Revankar, G. R., & Robins, R. K. (1984). Synthesis of 2′-deoxytubercidin, 2′-deoxyadenosine, and related 2′-deoxynucleosides via a novel direct stereospecific sodium salt glycosylation procedure. Journal of the American Chemical Society, 106(21), 6379-6382.
Robins, R. K. (1957). Potential purine antagonists. IX. Further studies of some 4,6-disubstituted pyrazolo[3,4-d]pyrimidines. Journal of the American Chemical Society, 79(23), 6407-6415.
Xu, S., Yao, H., Qiu, Y., Zhou, M., Li, D., Wu, L., ... & Xu, J. (2021). Discovery of novel polycyclic heterocyclic derivatives from evodiamine for the potential treatment of triple-negative breast cancer. Journal of Medicinal Chemistry, 64(23), 17346-17365.
Pavlov, V., Rodilla, V., & Lin, P. K. T. (2002). Growth, morphological and biochemical changes in oxa-spermine derivative-treated MCF-7 human breast cancer cells. Life sciences, 71(10), 1161-1173.
Audeh, M. W. (2014). Novel treatment strategies in triple-negative breast cancer: specific role of poly (adenosine diphosphate-ribose) polymerase inhibition. Pharmacogenomics and personalized medicine, 307-316.
Chiosis, G., Timaul, M. N., Lucas, B., Munster, P. N., Zheng, F. F., Sepp-Lorenzino, L., & Rosen, N. (2001). A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chemistry & biology, 8(3), 289-299.
Figueiredo, P., Costa, M., Pontes, O., Baltazar, F., & Proença, F. (2018). Adenine Derivatives: Promising Candidates for Breast Cancer Treatment. European Journal of Organic Chemistry, 2018(29), 3943-3956.
Dimopoulou, A., Manta, S., Parmenopoulou, V., Kollatos, N., Christidou, O., Triantakonstanti, V. V., ... & Komiotis, D. (2015). An easy microwave-assisted synthesis of C 8-alkynyl adenine pyranonucleosides as novel cytotoxic antitumor agents. Frontiers in Chemistry, 3, 21.
Jabłoński, A., Matczak, K., Koceva-Chyła, A., Durka, K., Steverding, D., Jakubiec-Krześniak, K., ... & Kowalski, K. (2017). Cymantrenyl-nucleobases: Synthesis, anticancer, antitrypanosomal and antimicrobial activity studies. Molecules, 22(12), 2220.
Kamper, C., Korpis, K., Specker, E., Anger, L., Neuenschwander, M., Bednarski, P. J., & Link, A. (2012). Sustainable synthesis and automated deposition: an accessible discovery screening library of fragment-like purines. Molecular diversity, 16, 541-551.
Proszenyak, A., Charnock, C., Hedner, E., Larsson, R., Bohlin, L., & Gundersen, L. L. (2007). Synthesis, Antimicrobial and Antineoplastic Activities for Agelasine and Agelasimine Analogs with a β‐Cyclocitral Derived Substituent. Archiv der Pharmazie: An International Journal Pharmaceutical and Medicinal Chemistry, 340(12), 625-634.
Džolić, Z., Krištafor, V., Cetina, M., Nagl, A., Hergold-Brundić, A., Mrvoš-Sermek, D., ... & Mintas, M. (2003). Synthesis, structural studies, and biological evaluation of some purine substituted 1-aminocyclopropane-1-carboxylic acids and 1-amino-1-hydroxymethylcyclopropanes. Nucleosides, Nucleotides and Nucleic Acids, 22(4), 373-389.
Prekupec, S., Svedružić, D., Gazivoda, T., Mrvoš-Sermek, D., Nagl, A., Grdiša, M., ... & Raić-Malić, S. (2003). Synthesis and biological evaluation of iodinated and fluorinated 9-(2-hydroxypropyl) and 9-(2-hydroxyethoxy) methyl purine nucleoside analogues. Journal of medicinal chemistry, 46(26), 5763-5772.
Mohammed, A. F., Andrei, G., Hayallah, A. M., Abdel-Moty, S. G., Snoeck, R., & Simons, C. (2019). Synthesis and anti-HSV activity of tricyclic penciclovir and hydroxybutylguanine derivatives. Bioorganic & Medicinal Chemistry, 27(6), 1023-1033.
Musumeci, D., Irace, C., Santamaria, R., Milano, D., Tecilla, P., & Montesarchio, D. (2015). Guanine-based amphiphiles: synthesis, ion transport properties and biological activity. Bioorganic & Medicinal Chemistry, 23(5), 1149-1156.
de Carvalho, O. V., Félix, D. M., de Mendonça, L. R., de Araújo, C. M. C. S., de Oliveira Franca, R. F., Cordeiro, M. T., ... & Pena, L. J. (2017). The thiopurine nucleoside analogue 6-methylmercaptopurine riboside (6MMPr) effectively blocks Zika virus replication. International journal of antimicrobial agents, 50(6), 718-725.
Winter, S. S., Dunsmore, K. P., Devidas, M., Wood, B. L., Esiashvili, N., Chen, Z., ... & Hunger, S. P. (2018). Improved survival for children and young adults with T-lineage acute lymphoblastic leukemia: results from the Children’s Oncology Group AALL0434 methotrexate randomization. Journal of Clinical Oncology, 36(29), 2926-2934.
Robins, R. K. (1964). Antitumor activity and structural relationships of purine derivatives and related compounds against neoplasms in experimental animals. Journal of Medicinal Chemistry, 7(2), 186-199.
Johnson, R. K., & Goldin, A. (1975). The clinical impact of screening and other experimental tumor studies. Cancer Treatment Reviews, 2(1), 1-31.
Johnson, R. K., & Goldin, A. (1975). The clinical impact of screening and other experimental tumor studies. Cancer Treatment Reviews, 2(1), 1-31.
Sharma, R. A., Bobek, M., & Bloch, A. (1975). Preparation and biological activity of some aminoacyl and peptidyl derivatives of 2'-amino-2'-deoxyuridine. Journal of Medicinal Chemistry, 18(9), 955-957.
Ariëns, E. J. (Ed.). (2013). Drug Design: Medicinal Chemistry: A Series of Monographs, Vol. 4 (Vol. 4). Elsevier.
Montgomery, J. A. (1982). Studies on the biologic activity of purine and pyrimidine analogs. Medicinal Research Reviews, 2(3), 271-308.
Samari, H. R., & Seglen, P. O. (1998). Inhibition of Hepatocytic Autophagy by Adenosine, Aminoimidazole-4-carboxamide Riboside, and N 6-Mercaptopurine Riboside: Evidence for involvement of amp-activated protein kinase. Journal of Biological Chemistry, 273(37), 23758-23763.
Hulpia, F., Noppen, S., Schols, D., Andrei, G., Snoeck, R., Liekens, S., ... & Van Calenbergh, S. (2018). Synthesis of a 3′-C-ethynyl-β-d-ribofuranose purine nucleoside library: discovery of C7-deazapurine analogs as potent antiproliferative nucleosides. European Journal of Medicinal Chemistry, 157, 248-267.
Kamal, A., Thao, L., Sensintaffar, J., Zhang, L., Boehm, M. F., Fritz, L. C., & Burrows, F. J. (2003). A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature, 425(6956), 407-410.
Llauger, L., He, H., Kim, J., Aguirre, J., Rosen, N., Peters, U., ... & Chiosis, G. (2005). Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90. Journal of medicinal chemistry, 48(8), 2892-2905.
Dymock, B., Barril, X., Beswick, M., Collier, A., Davies, N., Drysdale, M., ... & Wright, L. (2004). Adenine derived inhibitors of the molecular chaperone HSP90—SAR explained through multiple X-ray structures. Bioorganic & medicinal chemistry letters, 14(2), 325-328.
Maloney, A., & Workman, P. (2002). HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert opinion on biological therapy, 2(1), 3-24.
Chiosis, G., Lucas, B., Huezo, H., Solit, D., Basso, A., & Rosen, N. (2003). Development of purine-scaffold small molecule inhibitors of Hsp90. Current cancer drug targets, 3(5), 371-376.
Chang, Y. T., Wignall, S. M., Rosania, G. R., Gray, N. S., Hanson, S. R., Su, A. I., ... & Schultz, P. G. (2001). Synthesis and biological evaluation of myoseverin derivatives: microtubule assembly inhibitors. Journal of medicinal chemistry, 44(26), 4497-4500.
Piccoli, M., Palazzolo, G., Conforti, E., Lamorte, G., Papini, N., Creo, P., ... & Anastasia, L. (2012). The synthetic purine reversine selectively induces cell death of cancer cells. Journal of Cellular Biochemistry, 113(10), 3207-3217.
Wu, X., Ding, S., Ding, Q., Gray, N. S., & Schultz, P. G. (2002). A small molecule with osteogenesis-inducing activity in multipotent mesenchymal progenitor cells. Journal of the American Chemical Society, 124(49), 14520-14521.
Wu, X., Walker, J., Zhang, J., Ding, S., & Schultz, P. G. (2004). Purmorphamine induces osteogenesis by activation of the hedgehog signaling pathway. Chemistry & biology, 11(9), 1229-1238.
Chen, S., Zhang, Q., Wu, X., Schultz, P. G., & Ding, S. (2004). Dedifferentiation of lineage-committed cells by a small molecule. Journal of the American Chemical Society, 126(2), 410-411.
Sawyer, T. K., Shakespeare, W. C., Wang, Y., Sundaramoorthi, R., Huang, W. S., Metcalf, I. I. I., ... & Noehre, J. (2005). Protein phosphorylation and signal transduction modulation: chemistry perspectives for small-molecule drug discovery. Medicinal Chemistry, 1(3), 293-319.
Corbin, A. S., Demehri, S., Griswold, I. J., Wang, Y., Metcalf III, C. A., Sundaramoorthi, R., ... & Deininger, M. W. (2005). In vitro and in vivo activity of ATP-based kinase inhibitors AP23464 and AP23848 against activation-loop mutants of Kit. Blood, 106(1), 227-234.
Knockaert, M., Greengard, P., & Meijer, L. (2002). Pharmacological inhibitors of cyclin-dependent kinases. Trends in pharmacological sciences, 23(9), 417-425.
McClue, S. J., Blake, D., Clarke, R., Cowan, A., Cummings, L., Fischer, P. M., ... & Lane, D. P. (2002). In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R‐roscovitine). International journal of cancer, 102(5), 463-468.
Raynaud, F. I., Fischer, P. M., Nutley, B. P., Goddard, P. M., Lane, D. P., & Workman, P. (2004). Cassette dosing pharmacokinetics of a library of 2, 6, 9-trisubstituted purine cyclin-dependent kinase 2 inhibitors prepared by parallel synthesis. Molecular Cancer Therapeutics, 3(3), 353-362.
Roskoski Jr, R. (2004). Src protein–tyrosine kinase structure and regulation. Biochemical and biophysical research communications, 324(4), 1155-1164.
Roskoski Jr, R. (2015). Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacological research, 94, 9-25.
MacCallum, D. E., Melville, J., Frame, S., Watt, K., Anderson, S., Gianella-Borradori, A., ... & Green, S. R. (2005). Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II–dependent transcription and down-regulation of Mcl-1. Cancer research, 65(12), 5399-5407.
Fischer, P. M., & Gianella-Borradori, A. (2003). CDK inhibitors in clinical development for the treatment of cancer. Expert opinion on investigational drugs, 12(6), 955-970.
Zaidan, N. K., & Mohamed, W. M. (2016). Synthesis and Biological Effectiveness of Some new Azo Compounds as Derivatives of Nitrogen Bases. Baghdad science journal, 13(2), 0368-0368.
Ko, E. J., Ho Shin, Y., Hyun, H. N., Song, H. S., Hong, J. K., & Jeun, Y. C. (2019). Bio-sulfur pre-treatment suppresses anthracnose on cucumber leaves inoculated with Colletotrichum orbiculare. Mycobiology, 47(3), 308-318.
Slassi, S., Fix-Tailler, A., Larcher, G., Amine, A., & El-Ghayoury, A. (2019). Imidazole and Azo‐Based Schiff Bases Ligands as Highly Active Antifungal and Antioxidant Components. Heteroatom Chemistry, 2019(1), 6862170.
Thomson, J. M., & Lamont, I. L. (2019). Nucleoside analogues as antibacterial agents. Frontiers in microbiology, 10, 952.
Joshi, S., Bilgaiyan, P., & Pathak, A. (2017). Synthesis, spectroscopic characterization and antibacterial screening of novel Mannich bases of Ganciclovir. Arabian Journal of Chemistry, 10, S1180-S1187.
Gundersen, L. L., Nissen-Meyer, J., & Spilsberg, B. (2002). Synthesis and antimycobacterial activity of 6-arylpurines: the requirements for the N-9 substituent in active antimycobacterial purines. Journal of Medicinal Chemistry, 45(6), 1383-1386.
El Kouni, M. H. (2003). Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacology & Therapeutics, 99(3), 283-309.
Abrahamsen, M. S., Templeton, T. J., Enomoto, S., et al. (2004). Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science, 304(5669), 441-445.
Chaudhary, K., Darling, J. A., Fohl, M. L., et al. (2004). Purine salvage pathways in the apicomplexan Toxoplasma gondii. The Journal of Biological Chemistry, 279(30), 31221-31227.
Ullman, B., & Carter, D. (1999). Molecular and biochemical studies on the hypoxanthine-guanine phosphoribosyltransferases of the pathogenic haemoflagellates. International Journal of Parasitology, 27(2), 203-213.
Naguib, F. N., Iltzsch, M. H., El Kouni, M. M., Panzica, R. P., & El Kouni, M. H. (1995). Structure–activity relationships for the binding of ligands to xanthine or guanine phosphoribosyltransferase from Toxoplasma gondii. Biochemical Pharmacology, 50(10), 1685-1693.
Sommer, J. M., Ma, H., & Wang, C. C. (1999). Cloning, expression, and characterization of an unusual guanine phosphoribosyltransferase from Giardia lamblia. Molecular and Biochemical Parasitology, 78(1-2), 185-193.
Suchail, S., Sarciron, M. E. S., & Petavy, A. F. (1998). Purine metabolism in Echinococcus multilocularis. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 120(4), 633-637.
Doyle, P. S., Kanaani, J., & Wang, C. C. (1998). Hypoxanthine, guanine, xanthine phosphoribosyltransferase activity in Cryptosporidium parvum. Experimental Parasitology, 89(1), 9-15.
Bakkestuen, A. K., Gundersen, L. L., Langli, G., Liu, F., & Nolsøe, J. M. (2000). 9-Benzylpurines with inhibitory activity against Mycobacterium tuberculosis. Bioorganic & medicinal chemistry letters, 10(11), 1207-1210.
Pathak, A. K., Pathak, V., Seitz, L. E., Suling, W. J., & Reynolds, R. C. (2004). Antimycobacterial agents. 1. Thio analogues of purine. Journal of medicinal chemistry, 47(1), 273-276.
Bakkestuen, A. K., Gundersen, L. L., & Utenova, B. T. (2005). Synthesis, biological activity, and SAR of antimycobacterial 9-aryl-, 9-arylsulfonyl-, and 9-benzyl-6-(2-furyl) purines. Journal of medicinal chemistry, 48(7), 2710-2723.
Bakkestuen, A. K., Gundersen, L. L., Petersen, D., Utenova, B. T., & Vik, A. (2005). Synthesis and antimycobacterial activity of agelasine E and analogs. Organic & Biomolecular Chemistry, 3(6), 1025-1033.
Wang, J., Singh, U. S., Rawal, R. K., Sugiyama, M., Yoo, J., Jha, A. K., ... & Chu, C. K. (2011). Antiviral activity of novel 2′-fluoro-6′-methylene-carbocyclic adenosine against wild-type and drug-resistant hepatitis B virus mutants. Bioorganic & medicinal chemistry letters, 21(21), 6328-6331.
Haraguchi, K., Shimada, H., Kimura, K., Akutsu, G., Tanaka, H., Abe, H., ... & Balzarini, J. (2011). Synthesis of 4′-Ethynyl-2′-deoxy-4′-thioribonucleosides and Discovery of a Highly Potent and Less Toxic NRTI. ACS medicinal chemistry letters, 2(9), 692-697.
Ko, O. H., & Hong, J. H. (2004). Synthesis and biological evaluation of novel 2′, 3′, 4′‐triply branched carbocyclic nucleosides as potential antiviral agents. Archiv der Pharmazie: An International Journal Pharmaceutical and Medicinal Chemistry, 337(11), 579-586.
Ahn, H. S., Bercovici, A., Boykow, G., Bronnenkant, A., Chackalamannil, S., Chow, J., ... & Zhang, H. (1997). Potent tetracyclic guanine inhibitors of PDE1 and PDE5 cyclic guanosine monophosphate phosphodiesterases with oral antihypertensive activity. Journal of Medicinal Chemistry, 40(14), 2196-2210.
Hirota, K., Kazaoka, K., Niimoto, I., Kumihara, H., Sajiki, H., Isobe, Y., ... & Kawakami, H. (2002). Discovery of 8-hydroxyadenines as a novel type of interferon inducer. Journal of medicinal chemistry, 45(25), 5419-5422.
Reddy, K. R., Matelich, M. C., Ugarkar, B. G., Gómez-Galeno, J. E., DaRe, J., Ollis, K., ... & Erion, M. D. (2008). Pradefovir: a prodrug that targets adefovir to the liver for the treatment of hepatitis B. Journal of medicinal chemistry, 51(3), 666-676.
De Clercq, E. (2010). Antiretroviral drugs. Current Opinion in Pharmacology, 10(5), 507-515.
Chow, W. A., Jiang, C., & Guan, M. (2009). Anti-HIV drugs for cancer therapeutics: Back to the future? The Lancet Oncology, 10(1), 61-71.
Humer, J., Ferko, B., Waltenberger, A., Rapberger, R., Pehamberger, H., & Muster, T. (2008). Azidothymidine inhibits melanoma cell growth in vitro and in vivo. Melanoma Research, 18(5), 314-321.
Mattson, D. M., Ahmad, I. M., Dayal, D., Parsons, A. D., Aykin-Burns, N., Li, L., Orcutt, K. P., Spitz, D. R., Dornfeld, K. J., & Simons, A. L. (2009). Cisplatin combined with zidovudine enhances cytotoxicity and oxidative stress in human head and neck cancer cells via a thiol-dependent mechanism. Free Radical Biology and Medicine, 46(2), 232-237.
Kchour, G., Tarhini, M., Kooshyar, M. M., El Hajj, H., Wattel, E., Mahmoudi, M., Hatoum, H., Rahimi, H., Maleki, M., Rafatpanah, H., Rezaee, S. A., Yazdi, M. T., Shirdel, A., de Thé, H., Hermine, O., Farid, R., & Bazarbach, A. (2009). Phase 2 study of the efficacy and safety of the combination of arsenic trioxide interferon alpha and zidovudine in newly diagnosed chronic adult T-cell leukemia/lymphoma (ATL). Blood, 113(26), 6528-6532.
Vilček, J., & Jahiel, R. I. (1970). Action of interferon and its inducers against nonviral infectious agents. Archives of Internal Medicine, 126(1), 69-77.
Merigan, T. C., De Clercq, E., & Bausek, G. H. (1970). Nonviral interferon inducers. The Journal of General Physiology, 56(1), 57-75.
De Clercq, E. (2006). Interferon and its inducers—a never-ending story:“old” and “new” data in a new perspective. The Journal of infectious diseases, 194(Supplement_1), S19-S26.
Rhim, J. S., & Huebner, R. J. (1971). Comparison of the antitumor effect of interferon and interferon inducers. Proceedings of the Society for Experimental Biology and Medicine, 136(2), 524-529.
de Clercq, E. (1980). Interferon inducers. Virus Chemotherapy, 27, 251-287.
Brüning, A., Friese, K., Burges, A., & Mylonas, I. (2010). Tamoxifen enhances the cytotoxic effects of nelfinavir in breast cancer cells. Breast Cancer Research, 12(2), R45.
Perry, C. M. (2009). Emtricitabine/tenofovir disoproxil fumarate: In combination with a protease inhibitor in HIV-1 infection. Drugs, 69(7), 843-857.
Kamiya, N., Kubota, A., Iwase, Y., Sekiya, K., Ubasawa, M., & Yuasa, S. (2002). Antiviral activities of MCC-478, a novel and specific inhibitor of hepatitis B virus. Antimicrobial agents and chemotherapy, 46(9), 2872-2877.
Raboisson, P., Lugnier, C., Muller, C., Reimund, J. M., Schultz, D., Pinna, G., ... & Bourguignon, J. J. (2003). Design, synthesis and structure–activity relationships of a series of 9-substituted adenine derivatives as selective phosphodiesterase type-4 inhibitors. European journal of medicinal chemistry, 38(2), 199-214.
Cammarota, G., Ianiro, G., & Gasbarrini, A. (2014). Fecal microbiota transplantation for the treatment of Clostridium difficile infection: a systematic review. Journal of clinical gastroenterology, 48(8), 693-702.
Crawford, T., Huesgen, E., & Danziger, L. (2012). Fidaxomicin: a novel macrocyclic antibiotic for the treatment of Clostridium difficile infection. American Journal of Health-System Pharmacy, 69(11), 933-943.
Freeman, J., Bauer, M. P., Baines, S. D., Corver, J., Fawley, W. N., Goorhuis, B., ... & Wilcox, M. H. (2010). The changing epidemiology of Clostridium difficile infections. Clinical microbiology reviews, 23(3), 529-549.
McFarland, L. V., Beneda, H. W., Clarridge, J. E., & Raugi, G. J. (2007). Implications of the changing face of Clostridium difficile disease for health care practitioners. American journal of infection control, 35(4), 237-253.
Lucado, J., Gould, C., & Elixhauser, A. (2012). Clostridium difficile infections (CDI) in hospital stays, 2009.
Giacchello, I., Musumeci, F., D’Agostino, I., Greco, C., Grossi, G., & Schenone, S. (2021). Insights into RNA-dependent RNA polymerase inhibitors as antiinfluenza virus agents. Current Medicinal Chemistry, 28(6), 1068-1090.
Mayhoub, A. S. (2012). Hepatitis C RNA-dependent RNA polymerase inhibitors: a review of structure–activity and resistance relationships; different scaffolds and mutations. Bioorganic & medicinal chemistry, 20(10), 3150-3161.
Tian, L., Qiang, T., Liang, C., Ren, X., Jia, M., Zhang, J., ... & Liu, H. (2021). RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. European journal of medicinal chemistry, 213, 113201.
Pokhrel, R., Chapagain, P., & Siltberg-Liberles, J. (2020). Potential RNA-dependent RNA polymerase inhibitors as prospective therapeutics against SARS-CoV-2. Journal of medical microbiology, 69(6), 864-873.
Campagnola, G., Gong, P., & Peersen, O. B. (2011). High-throughput screening identification of poliovirus RNA-dependent RNA polymerase inhibitors. Antiviral research, 91(3), 241-251.
Koudriakova, T., Manouilov, K. K., Shanmuganathan, K., Kotra, L. P., Boudinot, F. D., Cretton-Scott, E., ... & Chu, C. K. (1996). In Vitro and in Vivo Evaluation of 6-Azido-2 ‘, 3 ‘-dideoxy-2 ‘-fluoro-β-d-arabinofuranosylpurine and N 6-Methyl-2 ‘, 3 ‘-dideoxy-2 ‘-fluoro-β-d-arabinofuranosyladenine as Prodrugs of the Anti-HIV Nucleosides 2 ‘-F-ara-ddA and 2 ‘-F-ara-ddI. Journal of medicinal chemistry, 39(23), 4676-4681.
Warren, T. K., Wells, J., Panchal, R. G., Stuthman, K. S., Garza, N. L., Van Tongeren, S. A., ... & Bavari, S. (2014). Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature, 508(7496), 402-405.
De Wilde, A. H., Snijder, E. J., Kikkert, M., & van Hemert, M. J. (2018). Host factors in coronavirus replication. Roles of host gene and non-coding RNA expression in virus infection, 1-42.
Singh, A. K., Singh, A., Singh, R., & Misra, A. (2020). Remdesivir in COVID-19: A critical review of pharmacology, pre-clinical and clinical studies. Diabetes & Metabolic Syndrome: Clinical Research & Reviews, 14(4), 641-648.
Malin, J. J., Spinner, C. D., Janssens, U., Welte, T., Weber-Carstens, S., Schälte, G., ... & Kluge, S. (2022). Key summary of German national treatment guidance for hospitalized COVID-19 patients: Key pharmacologic recommendations from a national German living guideline using an Evidence to Decision Framework (last updated 17.05. 2021). Infection, 50(1), 93-106.
Liang, C., Tian, L., Liu, Y., Hui, N., Qiao, G., Li, H., ... & Zhao, X. (2020). A promising antiviral candidate drug for the COVID-19 pandemic: A mini-review of remdesivir. European journal of medicinal chemistry, 201, 112527.
Ko, W. C., Rolain, J. M., Lee, N. Y., Chen, P. L., Huang, C. T., Lee, P. I., & Hsueh, P. R. (2020). Arguments in favour of remdesivir for treating SARS-CoV-2 infections. International journal of antimicrobial agents, 55(4), 105933.
Wang, Y., Zhang, D., Du, G., Du, R., Zhao, J., Jin, Y., ... & Wang, C. (2020). Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. The lancet, 395(10236), 1569-1578.
Wang, Y., Zhou, F., Zhang, D., Zhao, J., Du, R., Hu, Y., ... & Wang, C. (2020). Evaluation of the efficacy and safety of intravenous remdesivir in adult patients with severe COVID-19: study protocol for a phase 3 randomized, double-blind, placebo-controlled, multicentre trial. Trials, 21, 1-11.
Mohiuddin Chowdhury, A. T. M., Kamal, A., Abbas, K. U., Talukder, S., Karim, M. R., Ali, M. A., ... & He, S. (2022). Efficacy and outcome of remdesivir and tocilizumab combination against dexamethasone for the treatment of severe COVID-19: A randomized controlled trial. Frontiers in Pharmacology, 13, 690726.
Jachymek, M., Cader, A., Ptak, M., Witkiewicz, W., Szymański, A. G., Kotfis, K., ... & Szylińska, A. (2022). The value of clinical frailty scale (CFS) as a prognostic tool in predicting mortality in COVID-19—a retrospective cohort study. International Journal of Environmental Research and Public Health, 19(3), 1104.
Santoro, M. G., & Carafoli, E. (2021). Remdesivir: from Ebola to COVID-19. Biochemical and biophysical research communications, 538, 145-150.
Julander, J. G., Demarest, J. F., Taylor, R., Gowen, B. B., Walling, D. M., Mathis, A., & Babu, Y. S. (2021). An update on the progress of galidesivir (BCX4430), a broad-spectrum antiviral. Antiviral research, 195, 105180.
Aftab, S. O., Ghouri, M. Z., Masood, M. U., Haider, Z., Khan, Z., Ahmad, A., & Munawar, N. (2020). Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. Journal of translational medicine, 18, 1-15.
Avšič-Županc, T., Saksida, A., & Korva, M. (2019). Hantavirus infections. Clinical Microbiology and Infection, 21, e6-e16.
US National Institutes of Health, & Phase, A. (1). study to evaluate the safety, tolerability and pharmacokinetics of BCX4430. NCT02319772.
Sarkar, C., Mondal, M., Torequl Islam, M., Martorell, M., Docea, A. O., Maroyi, A., ... & Calina, D. (2020). Potential therapeutic options for COVID-19: current status, challenges, and future perspectives. Frontiers in pharmacology, 11, 572870.
Aftab, S. O., Ghouri, M. Z., Masood, M. U., Haider, Z., Khan, Z., Ahmad, A., & Munawar, N. (2020). Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. Jo
urnal of translational medicine, 18, 1-15.
Avšič-Županc, T., Saksida, A., & Korva, M. (2019). Hantavirus infections. Clinical Microbiology and Infection, 21, e6-e16.
Kowalska, A., Latocha, M., & Pluta, K. (2015). Synthesis and anticancer activity of thiosubstituted purines. Medicinal Chemistry Research, 24, 3107-3116.
Kowalska, A., Pluta, K., & Latocha, M. (2018). Synthesis and anticancer activity of multisubstituted purines and xanthines with one or two propynylthio and aminobutynylthio groups. Medicinal Chemistry Research, 27, 1384-1395.
Schabel Jr, F. M. (1962). Experimental evaluation of purine antagonists as anticancer agents. Cancer Chemother. Rep, 16, 37-42.
Tuncbilek, M., Kucukdumlu, A., Guven, E. B., Altiparmak, D., & Cetin-Atalay, R. (2018). Synthesis of novel 6-substituted amino-9-(β-d-ribofuranosyl) purine analogs and their bioactivities on human epithelial cancer cells. Bioorganic & medicinal chemistry letters, 28(3), 235-239.
Yoon, J. S., Jarhad, D. B., Kim, G., Nayak, A., Zhao, L. X., Yu, J., ... & Jeong, L. S. (2018). Design, synthesis and anticancer activity of fluorocyclopentenyl-purines and–pyrimidines. European Journal of Medicinal Chemistry, 155, 406-417.
Huang, L. H., Xu, H. D., Yang, Z. Y., Zheng, Y. F., & Liu, H. M. (2014). Synthesis and anticancer activity of novel C6-piperazine substituted purine steroid–nucleosides analogues. Steroids, 82, 1-6.
Student Names
- Noor Fatime
- Maryam Saeed
- Ayesha shafique
